MENU
CH | CHF

CH | CHF
-
- All Pipettes, Dispensers & Automated Liquid Handlers
- Mechanical Pipettes
- Electronic Pipettes
- Multi-Channel Pipettes
- Positive Displacement Pipettes & Dispensers
- Bottle-Top Dispensers
- Pipette Controllers
- Dispenser & Pipette Accessories
- Automated Pipetting
- Automation Consumables
- Automation Accessories
- Liquid Handler & Pipette Services
Sorry, we couldn't find anything on our website containing your search term.
Sorry, we couldn't find anything on our website containing your search term.
How Much Template Will I Add to My PCR Reaction?
Lab Academy
- Molecular Biology
- Amplification & PCR
- Photometry
- Photometers
- Essay
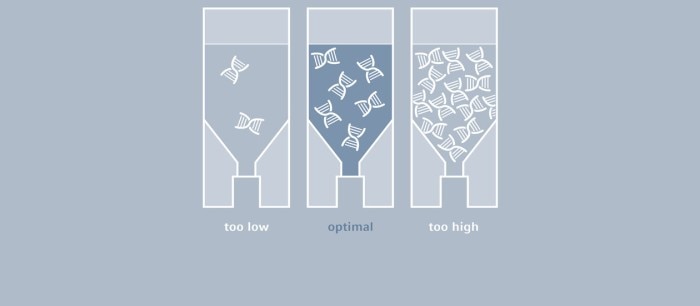
Even though in theory, one molecule of the template would be sufficient, considerably larger amounts of DNA are typically used for a classic PCR, for example, up to 1 µg of genomic mammalian DNA and as little as 1 pg of plasmid DNA (1). The optimal amount depends largely on the number of copies of the target sequence, as well as on its complexity.
If very little template is used, a corresponding increase in the number of amplification cycles will be needed to obtain a sufficient amount of product. A Taq polymerase that is used for most PCR experiments does not feature a correction function (3'-5' exonuclease activity); thus, errors occurring during amplification cannot be corrected. The higher the number of cycles, the more prevalent the amplification of flawed product will be. If, on the other hand, the amount of template is too high, the probability of primers annealing to other (not one hundred percent complimentary) sequences, as well as the formation of primer dimers, will increase, which will result in the amplification of by-products. In many cases, DNA is isolated from cell cultures or from microorganisms and subsequently used as a PCR template. Following purification, it is necessary to determine the concentration of the DNA to be able to define the volume that is required for the PCR setup. While agarose gel electrophoresis may serve to provide an estimate, this method is far from accurate. UV-Vis spectrophotometry has been established as the gold standard for the quantification of nucleic acids; this direct and therefore easy and quick method measures absorbance of the sample at 260 nm, and concentration is calculated with the help of a conversion factor.
Read more
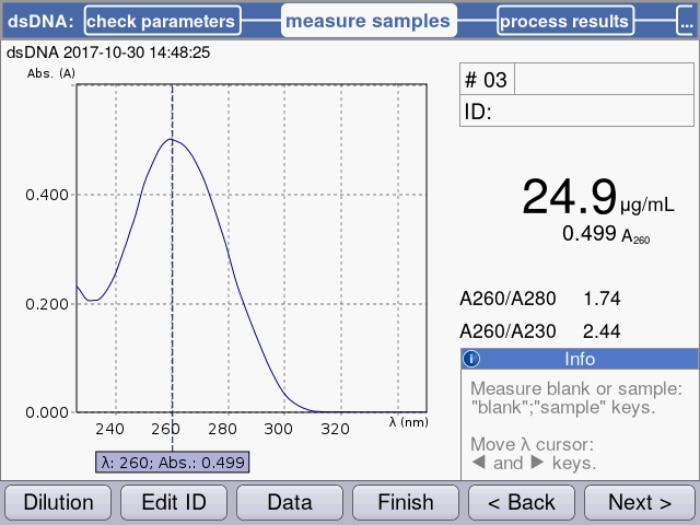
Figure 1: Peak maximum at 260 nm is used by the spectrophotometer to calculate the corresponding dsDNA concentration with the implemented specific factor of 50 µg/mL for dsDNA.
If the DNA concentration is very low, however (< 1 µg/mL dsDNA), or if it is contaminated with substances that also absorb in the 260 nm range (e. g. RNA, protein, salts), this method will reach its limitations. In the case of very low concentrations, the readings will soon become too inaccurate to be of use, and contaminations will lead to (sometimes enormous) overestimation of the actual value. In this case, quantification using fluorescence may present an alternative. This technique is based on the use of a fluorescent dye (e.g. PicoGreen®) that binds specifically to dsDNA only the complex comprising nucleic acid and dye is excited by the light, and it will subsequently emit light of a slightly higher wavelength. Here, the intensity of the fluorescent signal is proportional to the amount of DNA, and for determining the concentration it is evaluated in relation to a standard curve (Figure 2). The advantages of this method rest on the specificity of the bond, which excludes the external influences introduced by contamination, as well as on the resulting ability to detect very low concentrations of DNA (e.g. 1 ng/mL for PicoGreen). The suitability of either method depends mainly on sample concentration and purity; in many cases it may even be advisable to apply both methods in parallel (2).
Read more
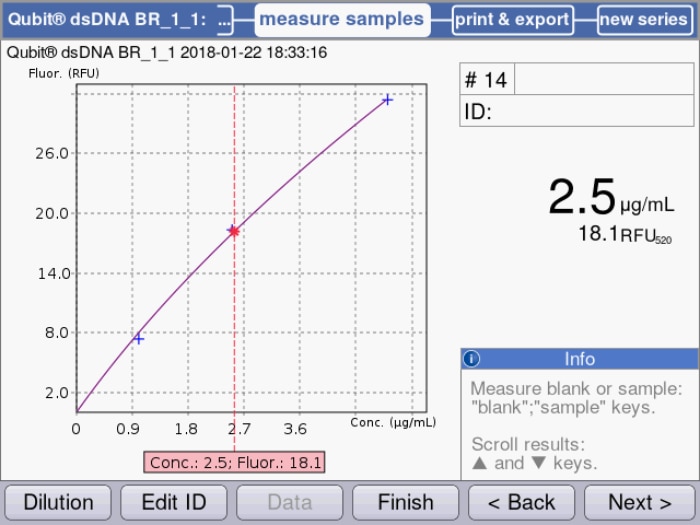
Figure 2: Standard curve based on quadratical regression of measured samples is used for the calculation of a dsDNA sample solution.
References:
[1] Sambrook, Fritsch, Maniatis (1989). Molecular Cloning: A laboratory manual. CSH Laboratory Press.
[2] Weiss N. Absorbance or fluorescence: How to quantify nucleic acids in the best way?. Eppendorf White Paper 40: www.eppendorf.com
PicoGreen® is a registered trademark of Molecular Probes, Inc. Corporation, Eugene, OR, USA.
[1] Sambrook, Fritsch, Maniatis (1989). Molecular Cloning: A laboratory manual. CSH Laboratory Press.
[2] Weiss N. Absorbance or fluorescence: How to quantify nucleic acids in the best way?. Eppendorf White Paper 40: www.eppendorf.com
PicoGreen® is a registered trademark of Molecular Probes, Inc. Corporation, Eugene, OR, USA.
Read more

Application Note
PDF
Application Note
PDF
Protocol
PDF
Application Note
PDF
White Paper
PDF